Example problem
- 1. Introduction / Modelling Scenario
- 2. Retrieve experimental kinetic data from the database SABIO-RK
- 3. Necessary model parameters and units adjustments
- 4. Model simulation
- 4. A use case for parameter estimation based on protein structural information with qPIPSA can be found here
1. Introduction / Modelling Scenario
Problem: Set up and simulate a model comprising the first two steps of glycolysis in human hepatocytes.
The file resulting from this use case can be loaded directly into SYCAMORE under 'Example models' or downloaded.
2. Retrieve experimental kinetic data from the database SABIO-RK
Since human data is rare, typically this will return only a small number. Therefore, we include Rattus norvegicus into search. For this example, we also use the corresponding PubMedID's for this search request.
Paste the following term into the search field:
d-glucose 6-phosphate AND Organism:("homo sapiens" OR "rattus norvegicus") AND Tissue:"liver" AND PubMedID:("1435758" OR "14988235")
You then submit your search by pressing the respective button.

The result page displays the reactions found that fullfill your search criteria. Press the blue arrow on the left to see more details.
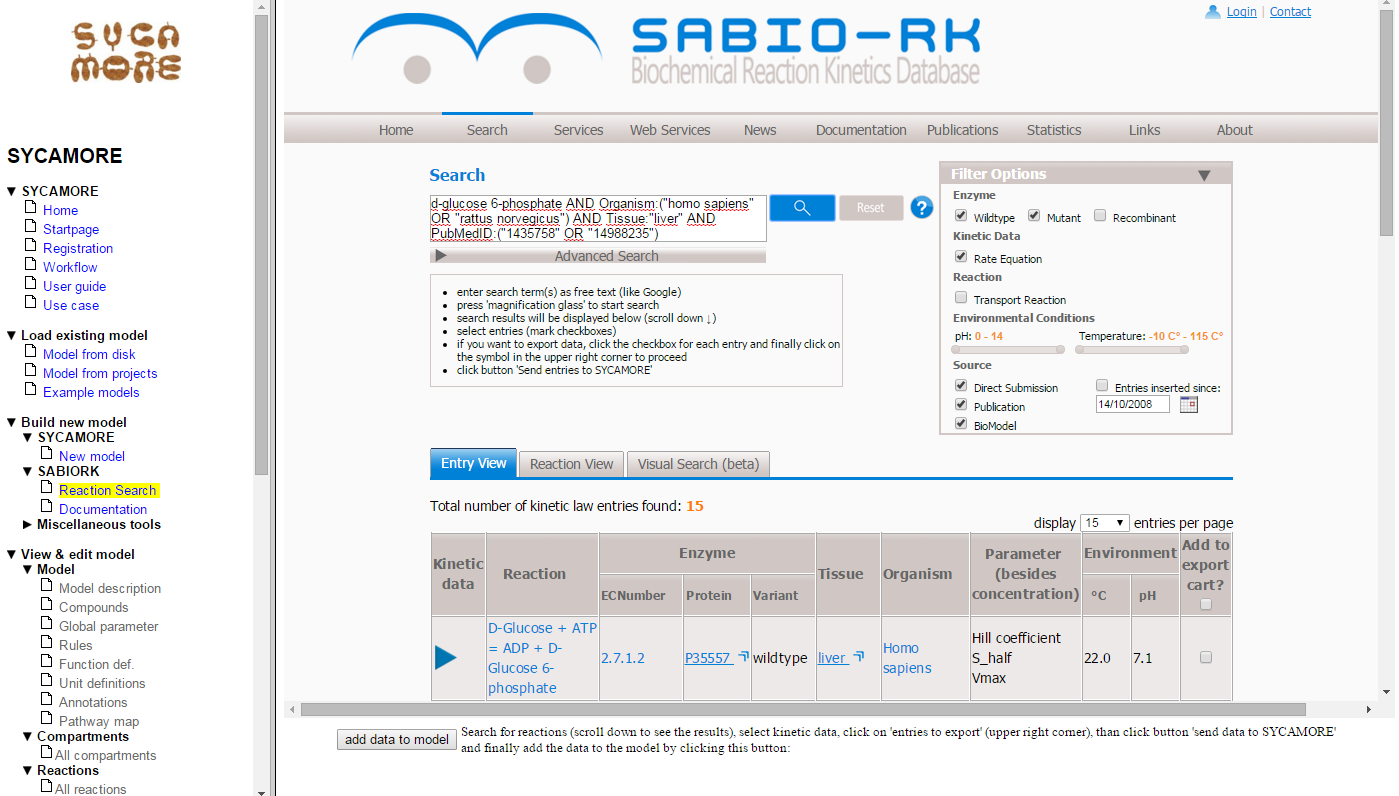
You now select those reactions that should be part of your model, in this case the first two steps of glycolysis. Please be aware that there might be several entries for each reactions depending on the level of detail describing the reactants given in the original publication. So, you might want to select several reactions for each step of glycolysis (e.g. reactions containing Glucose, D-Glucose and alpha-D-Glucose). Here we select kinetics data for the reactions:
- D-Glucose + ATP <=> ADP + D-Glucose 6-phosphate (Entry ID: 2580)
- D-Glucose 6-phosphate <=> D-Fructose 6-phosphate (Entry ID: 2616)
To select the data for export, click on the checkboxes on the right side for the entries 2580 and 2616.
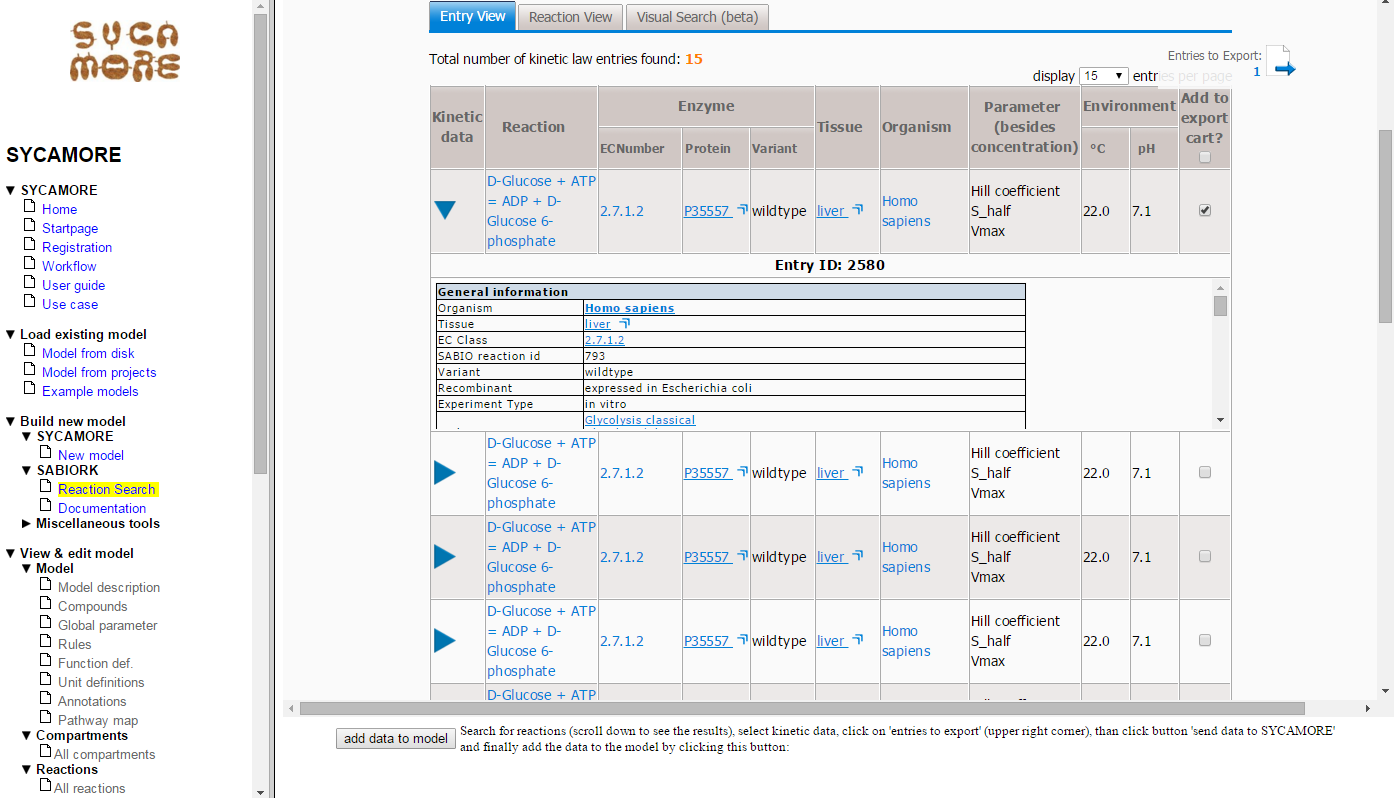
Thereafter, you can view the list of selected data by clicking on "Entries to Export (2)".
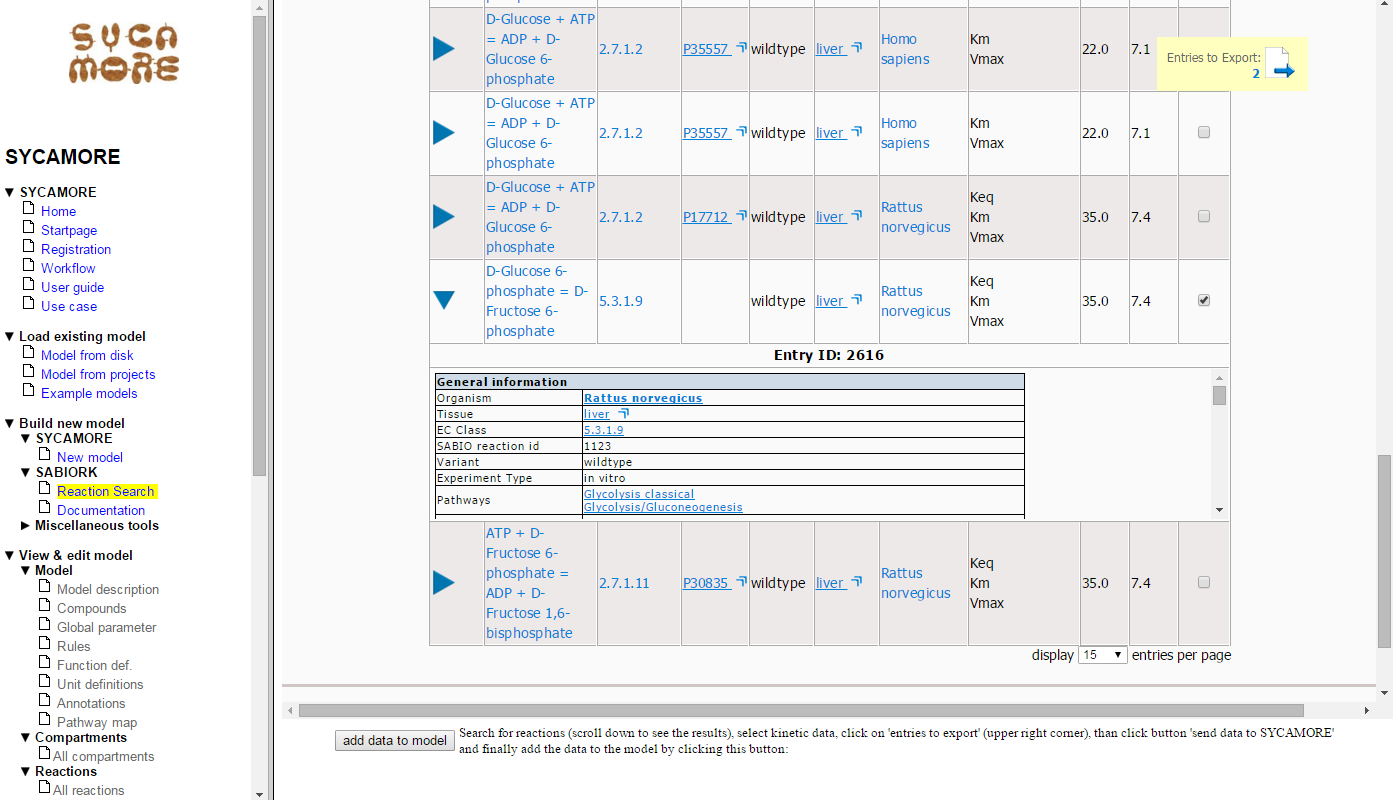
Here the selected entries are summarized. Press the button "Send Data to SYCAMORE".

The selected data are displayed in detail within the SYCAMORE context.
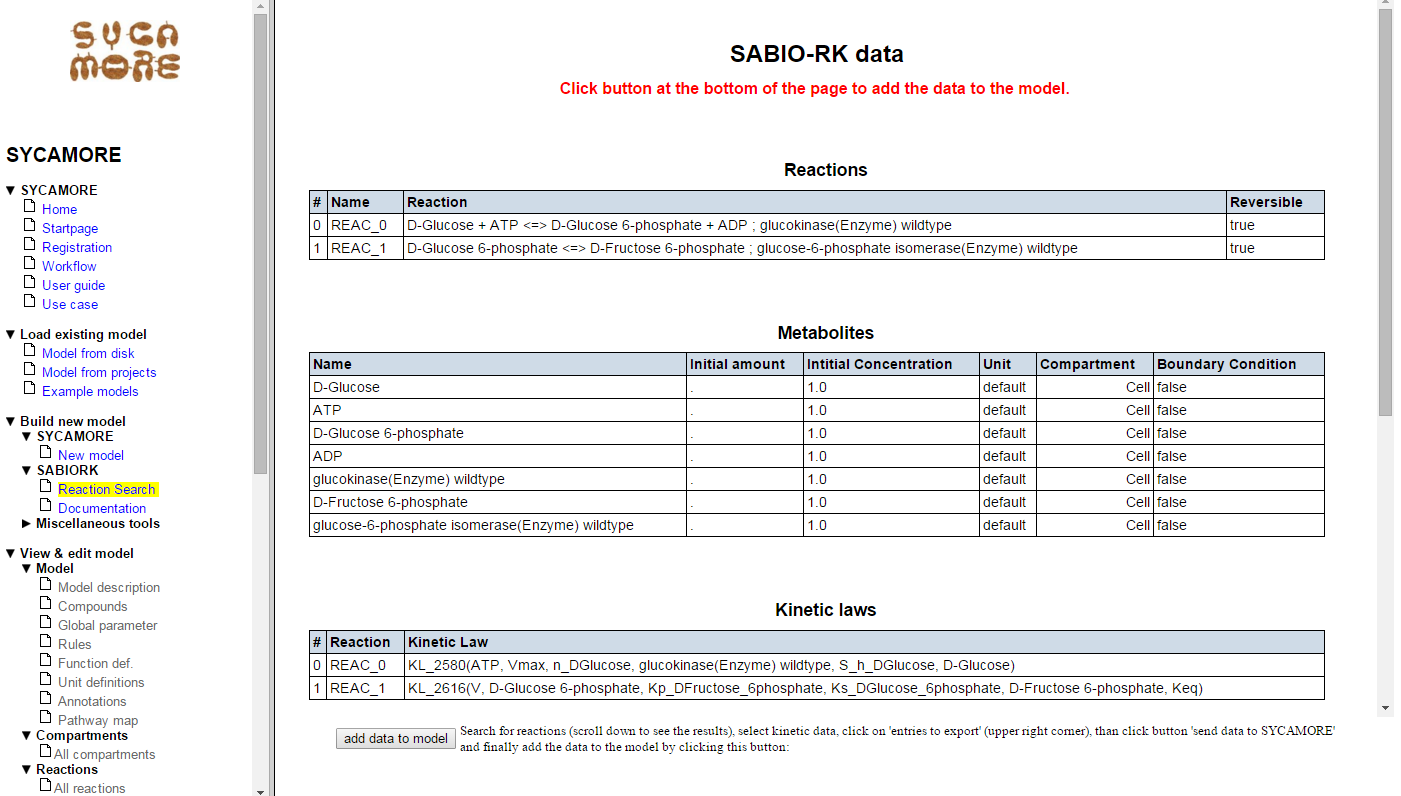
Now press the button "Add data to model". All data are now imported into SYCAMORE. The corresponding reactions are displayed on the left navigation panel as "reaction_0" and "reaction_1". By adding the data to the model you complete this first step.
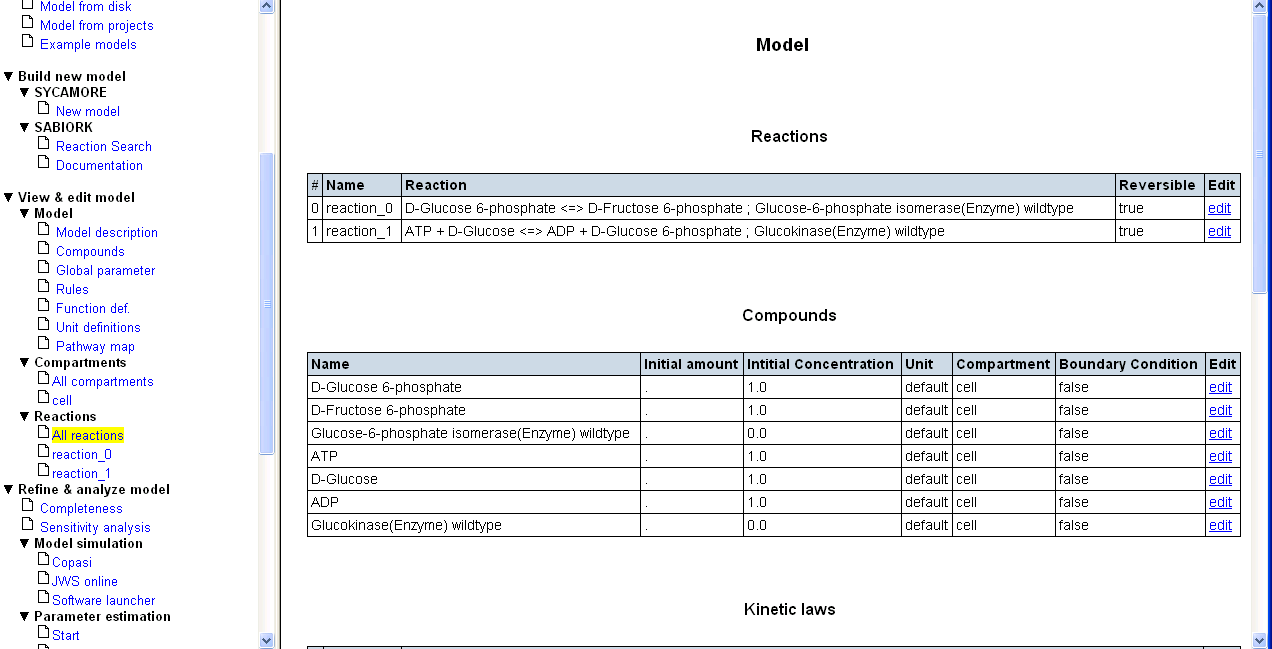
In this use case example, experimentally determined kinetic parameters were found. In cases where no such relevant data are retrieved, or data are only available for other species, you can start an estimate of enzymatic kinetic parameters using a structure-based modeling protocol (qPIPSA). A description for a parameter estimation based on protein structural information with qPIPSA can be found here.
3. Necessary model parameters and units adjustments
a) The compound names associated with the reactions have to be made consistent. As pointed out above depending on the literature compound names will differ e.g. in the chemical details. In order to make a consistent model you have to adjust them such that a single compound is not described by different names.
b) You should assign initial concentrations to your metabolites if you want to perform simulations later on. As a first guess you might check databases for typical concentrations of the respective metabolites in the tissue of interest or you just make an estimate.
c) You have to make units consistent. All units, e.g. with respect to time or mass should be the same within the model. That means you might have to transfer constants from one unit to another.
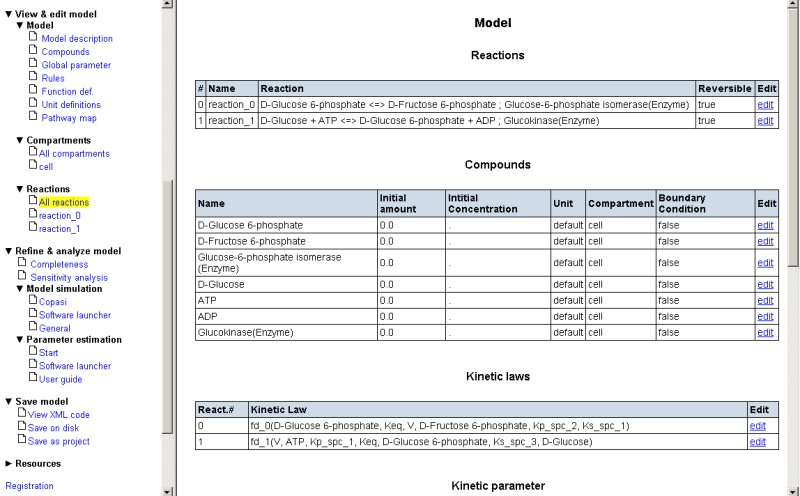
For the latter purpose you might have to adjust your units to the default units that you find under “unit definitions”. Later on, this might be automated, but right now, this has to be done manually.
One word of caution with respect to Vmax as found in SABIO-RK. Please be aware that these numbers represent values from literature which in turn usually represent in vitro experiments. Since Vmax is the product of the enzyme concentration times the kcat, with the enzyme concentration being the one in the test tube, this number will usually be not very close to the system you are interested in!
Rename the reactions (press 'edit' on the right hand site, than rename the reaction followed by 'update values' at the bottom of that page):
- 'reaction_0' to 'Glucokinase'
- 'reaction_1' to 'Isomerase'
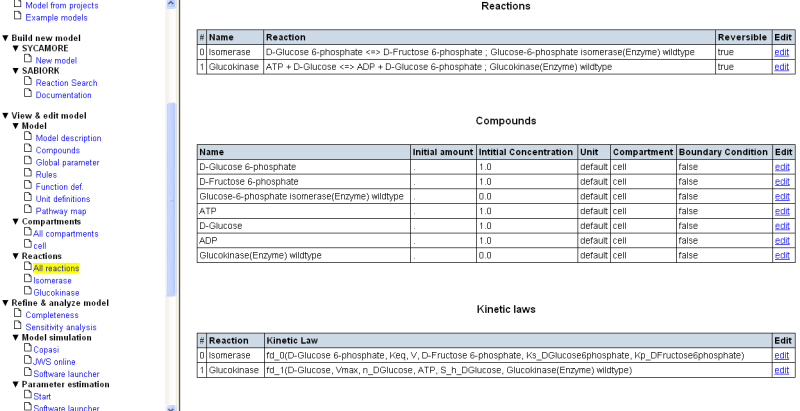
As a final step in setting up your draft model, you will likely want to make the system open. For this purpose you have to define an influx for Glucose and an efflux for Fructose-6-phosphate. This can be achieved by pressing the “add reaction” button on the bottom of the reaction list and defining two new reactions.
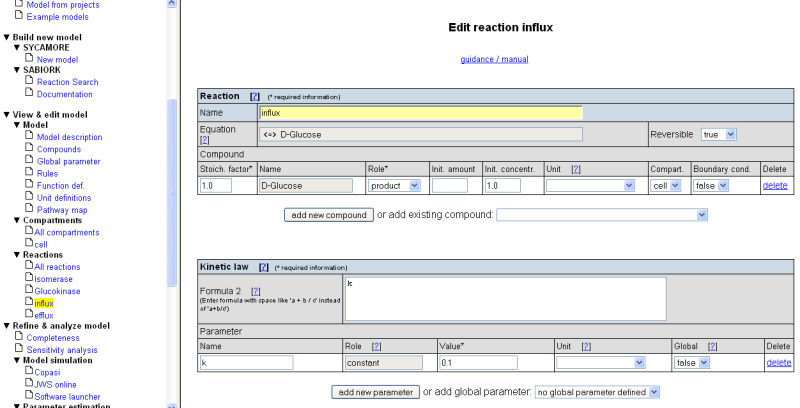
For the influx reaction fill in the following values:
- Reaction name: 'Influx'
- Select compound 'D-Glucose' from the selection list 'add existing compound'
- Specify the role of D-Glucose as 'product'
- Set the initial concentration to '1'
- Define a parameter 'k' for this reaction, set the value to '0.1'
- As kinetic law, enter 'k'
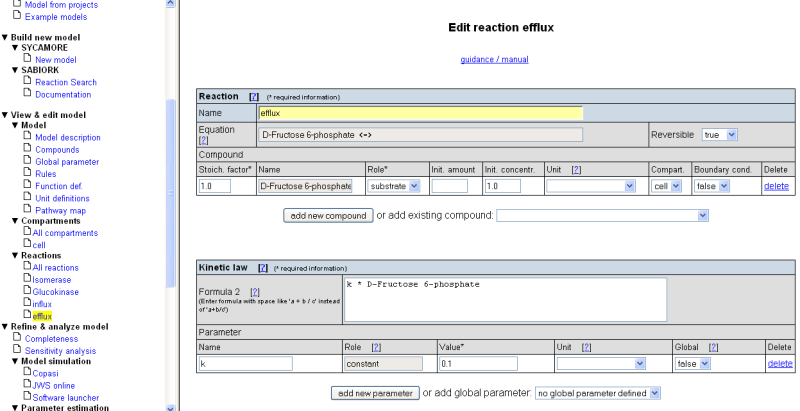
For the efflux reaction fill in the following values:
- Reaction name: 'Efflux'
- Select compound 'D-Fructose 6-phosphate' from the selection list 'add existing compound'
- Specify the role of D-Fructose 6-phosphate as 'substrate'
- Set the initial concentration to '1'
- Define a parameter 'k' for this reaction, set the value to '0.1'
- As kinetic law, enter 'k * D-Fructose 6-phosphate'
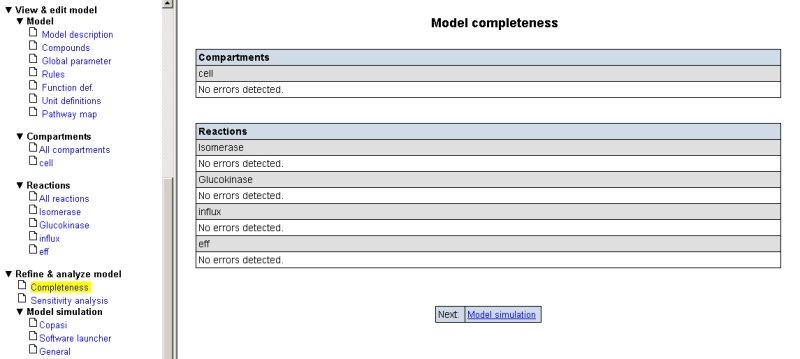
Now, you can check your draft model for completeness by selecting the respective option in the left panel. SYCAMORE will tell you if your model is complete with respect to kinetics and/or parameters.
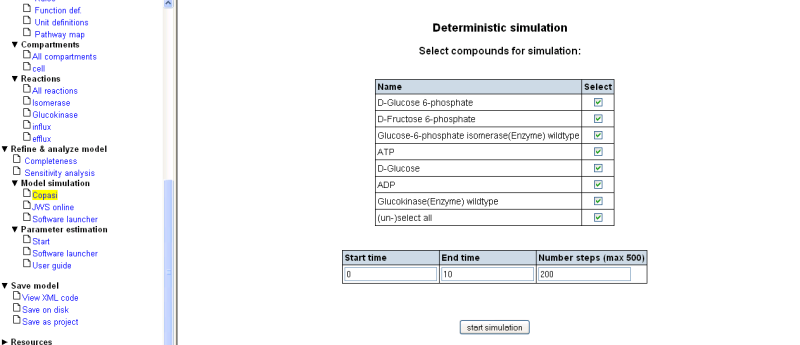
4. Model simulation
- Select checkbox 'select all'
- Set the 'End time' to 1.0
- Start simulation
You will get the following result:
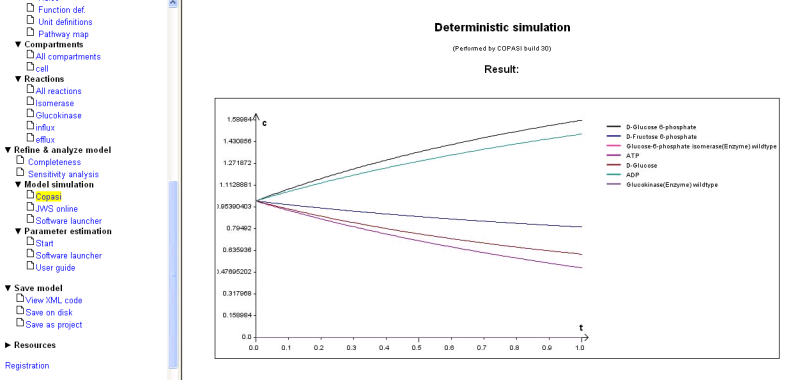
Please bear in mind that this quick simulation cannot substitute for a thourough simulation study with a tool of your choice. However, we want to provide you with the possibility to get a straightforward check of you model even before you go to more sophisticated analysis.
An alternative way to simulate the model is provided by using SYCAMORE's local software launcher. By clicking on the link 'Download software launcher', a Java Web Start Applikation is started that enables the simulation of models using either COPASI or ProMoT.
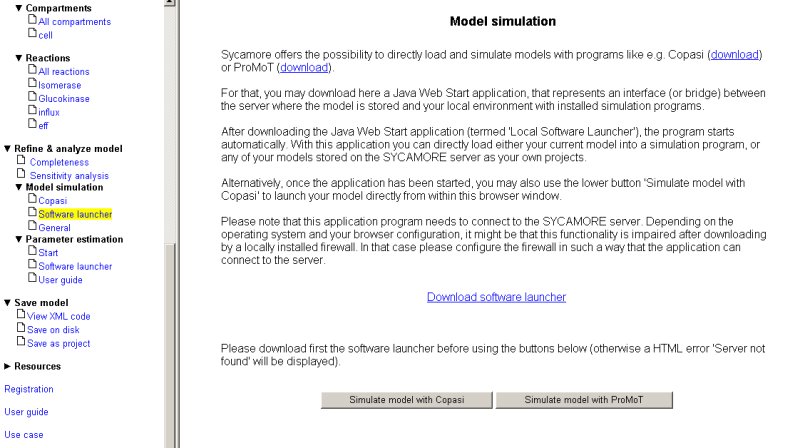
After launching the application, you have to set the path for the executables for COPASI and ProMoT, the path information is than stored on your disk, thus you do not have to re-enter this information every time you start the software launcher.
Additionally, you may provide your username and password used for the storage of your projects on the SYCAMORE server.
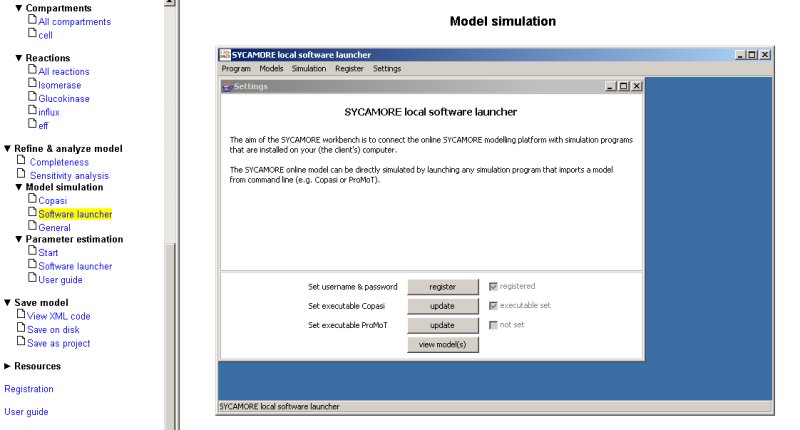
By pressing the button 'view models', a new screen appears were the name of your currently loaded model is displayed on the lower panel and - in case that you have registered before and that you store models on the SYCAMORE server - those models are shown on the upper panel.
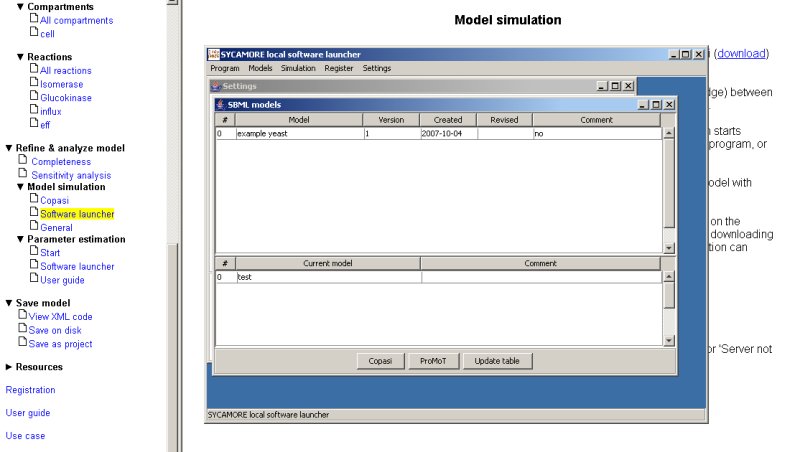
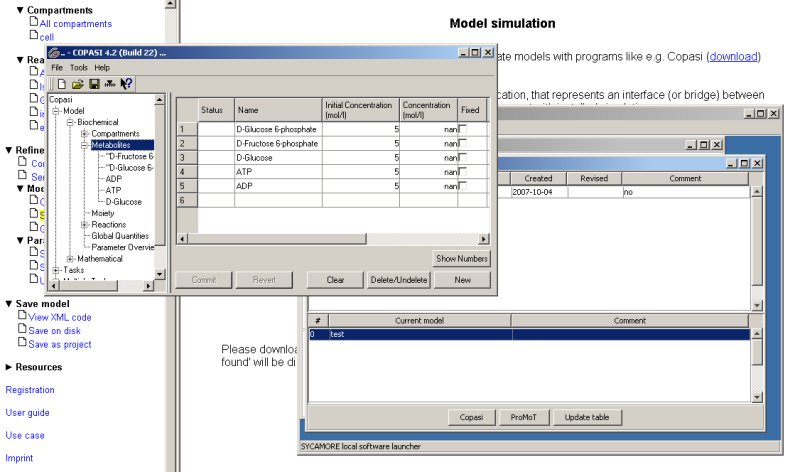
When you select any of the models displayed followed by pressing the button Copasi or ProMoT, the model is directly loaded by those simulation programs and can be simulated immediatly.
4. Parameter estimation based on protein structural information
(Last updated November 2015)